Thalassemia minor is generally considered a clinically asymptomatic condition with mild anemia. The hypercoagulable state and increased risk of thromboembolic events, including deep vein thrombosis (DVT), are more commonly associated with more severe forms of thalassemia, such as thalassemia intermedia and thalassemia major.
The literature indicates that patients with β-thalassemia intermedia and major are at a higher risk for thromboembolic events due to several factors, including chronic platelet activation, abnormal red blood cell membranes, and endothelial cell activation.[1-5] These complications are particularly pronounced in splenectomized patients and those not receiving regular transfusions.[6-7]
However, there is no strong evidence to suggest that individuals with thalassemia minor have a significantly increased risk of clotting or DVT. Thalassemia minor typically does not exhibit the same degree of ineffective erythropoiesis, hemolysis, or other pathophysiological changes that contribute to a hypercoagulable state in more severe forms of the disease.
To illustrate the pathophysiological and clinical manifestations of β-thalassemia, including the hypercoagulable state seen in more severe forms, the following figure from the New England Journal of Medicine is relevant:
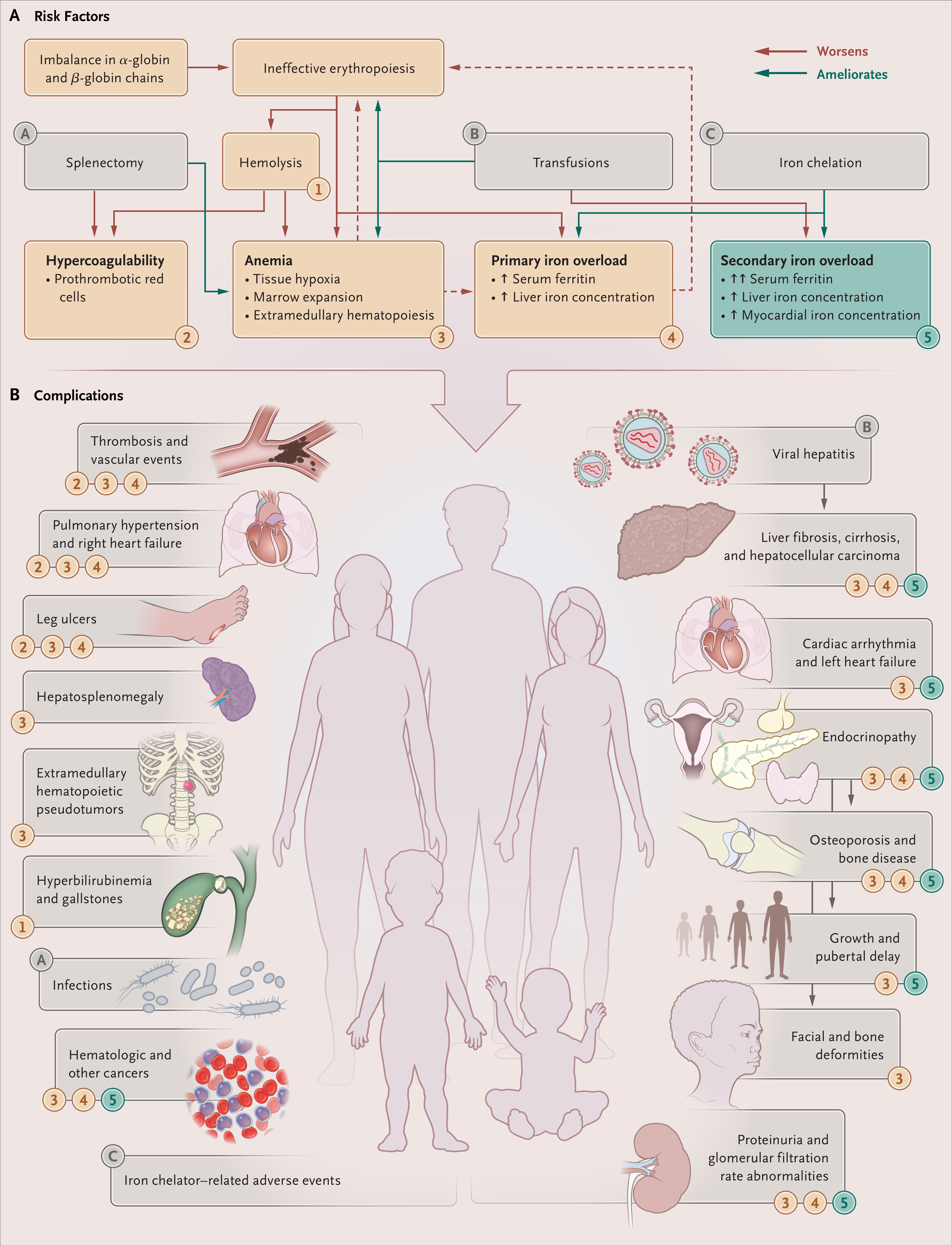
Figure 2. Pathophysiological and Clinical Manifestations of β-Thalassemia.
β-Thalassemias. N Engl J Med. February 25, 2021.
Used under license from The New England Journal of Medicine.
In summary, thalassemia minor is not associated with an increased risk of clotting or DVT. The hypercoagulable state and thromboembolic complications are primarily concerns in thalassemia intermedia and major.